Now, let's move on to actually defining the training models:
- First, make an empty list, in which we will append the KNN model.
- Enter the KNeighborsClassifier function and explore the number of neighbors.
- Start with n_neighbors = 5, and play around with the variable a little, to see how it changes our results.
- Next, we will add our models: the SVM and the SVC. We will evaluate each model, in turn.
- The next step will be to get a results list and a names list, so that we can print out some of the information at the end.
- We will then perform a for loop for each of the models defined previously, such as name or model in models.
- We will also do a k-fold comparison, which will run each of these a couple of times, and then take the best results. The number of splits, or n_splits, defines how many times it runs.
- Since we don't want a random state, we will go from the seed. Now, we will get our results. We will use the model_selection function that we imported previously, and the cross_val_score.
- For each model, we'll provide training data to X_train, and then y_train.
- We will also add the specification scoring, which was the accuracy that we added previously.
- We will also append results, name, and we will print out a msg. We will then substitute some variables.
- Finally, we will look at the mean results and the standard deviation.
- A k-fold training will take place, which means that this will be run 10 times. We will receive the average result and the average accuracy for each of them. We will use a random seed of 8, so that it is consistent across different trials and runs. Now, press Shift + Enter. We can see the output in the following screenshot:

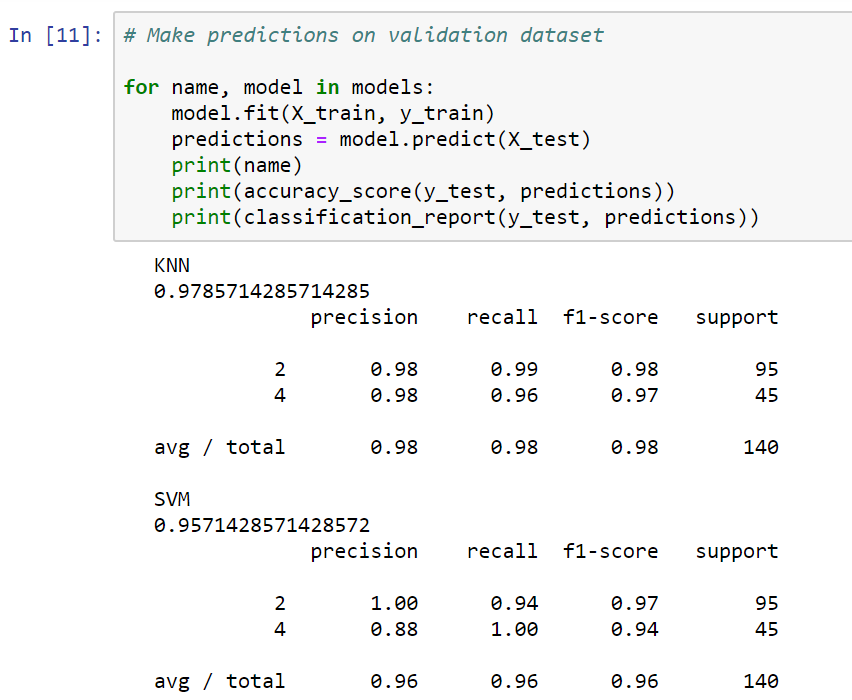
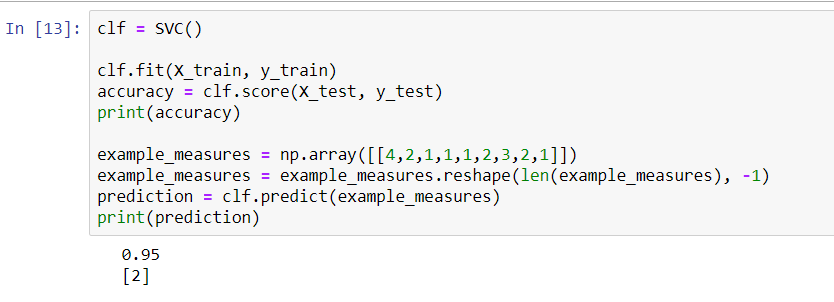
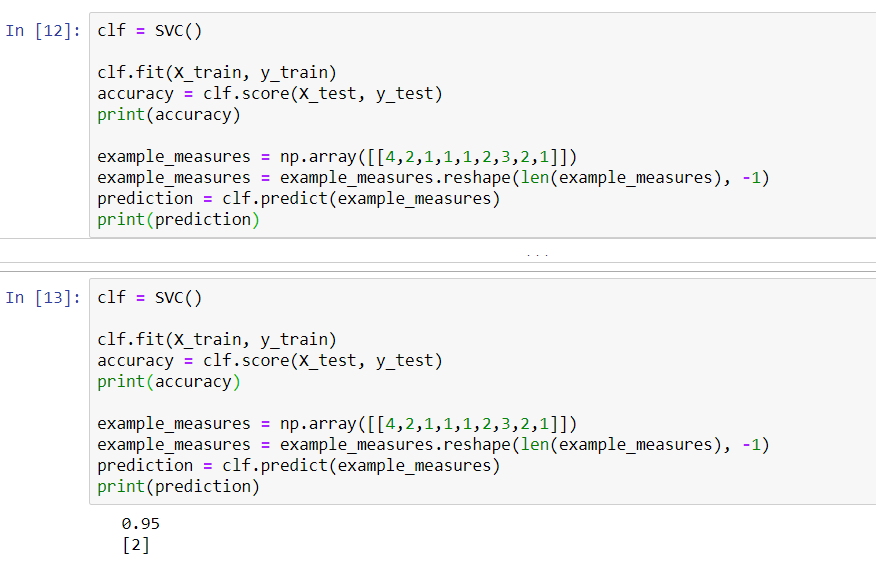
In this case, our KNN narrowly beats the SVC. We will now go back and make predictions on our validation set, because the numbers shown in the preceding screenshot just represent the accuracy of our training data. If we split up the datasets differently, we'll get the following results:
However, once again, it looks like we have pretty similar results, at least with regard to accuracy, on the training data between our KNN and our support vector classifier. The KNN tries to cluster the different data points into two groups: malignant and benign. The SVM, on the other hand, is looking for the optimal separating hyperplane that can separate these data points into malignant cells and benign cells.