






















































Models and data
Machine learning models work with data. They create associations, find out relationships, discover patterns, generate new samples, and more, working with well-defined datasets, which are homogenous collections of data points (for example, observations, images, or measures) related to a specific scenario (for example, the temperature of a room sampled every 5 minutes, or the weights of a population of individuals)
Unfortunately, sometimes the assumptions or conditions imposed on machine learning models are not clear, and a lengthy training process can result in a complete validation failure. We can think of a model as a gray box (some transparency is guaranteed by the simplicity of many common algorithms), where a vectoral input X extracted from a dataset is transformed into a vectoral output Y:

Schema of a generic model parameterized with the vector 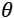 and its relationship with the real world
and its relationship with the real world
In the preceding diagram, the model has been represented by a function that depends on a set of parameters defined by the vector . The dataset is represented by data extracted from a real-world scenario, and the outcomes provided by the model must reflect the nature of the actual relationships. These conditions are very strong in logic and probabilistic contexts, where the inferred conditions must reflect natural ones.
For our purposes, it's necessary to define models that:
- Mimic animal cognitive functions
- Learn to produce outcomes that are compatible with the environment, given a proper training set
- Learn to overcome the boundaries of the training set, by outputting the correct (or the most likely) outcome when new samples are presented
The first point is a crucial element in the AI debate. As pointed out by Darwiche (in Darwiche A., Human-Level Intelligence or Animal-Like Abilities?, Communications of the ACM, Vol. 61, 10/2018), the success of modern machine learning is mainly due to the ability of deep neural networks to reproduce specific cognitive functions (for example, vision or speech recognition). It's obvious that the outcomes of such models must be based on real-world data and, moreover, that they must possess all the features of the outcomes generated by the animals whose cognitive functions we are trying to reproduce.
We're going to analyze these properties in detail. It's important to remember that they're not simple requirements, but rather the pillars that guarantee the success or the failure of an AI application in a production environment (that is, outside of the golden world of limited and well-defined datasets).
In this section, we're only considering parametric models, although there's a family of algorithms that are called non-parametric because they're only based on the structure of the data; we're going to discuss some of them in upcoming chapters.
The task of a parametric learning process is to find the best parameter set that maximizes a target function, the value of which is proportional to the accuracy of the model, given specific input X and output Y datasets (or proportional to the error, if we're trying to minimize the error). This definition isn't very rigorous, and we'll improve it in the following sections; however, it's useful as a way to introduce the structure and the properties of the data we're using, in the context of machine learning.
Structure and properties of the datasets
The first question to ask is: What are the natures of X and Y? A machine learning problem is focused on learning abstract relationships that allow a consistent generalization when new samples are provided. More specifically, we can define a stochastic data generating process with an associated joint probability distribution:

The process pdata represents the broadest and most abstract expression of the problem. For example, a classifier that must distinguish between male and female portraits will be based on a data generating process that theoretically defines the probabilities of all possible faces, with respect to the binary attribute male/female. It's clear that we can never work directly with pdata; it's only possible to find a well-defined formula describing pdata in a few limited cases (for example, the distribution of all images belonging to a dataset).
Even so, it's important for the reader to consider the existence of such a process, even when the complexity is too high to allow any direct mathematical modeling. A machine learning model must consider this kind of abstraction as a reference.
Limited Sample Populations
In many cases, we cannot derive a precise distribution and we're forced to work with a limited population of actual samples. For example, a pharmaceutical experiment is aimed to understand the effectiveness of a drug on human beings. Obviously, we cannot test the drug on every single individual, nor we can imagine including all dead and future people. Nevertheless, the limited sample population must be selected carefully, in order to represent the underlying data generating process. That is, all possible groups, subgroups, and reactions must be considered.
Since this is generally impossible, it's necessary to sample from a large population. Sampling, even in the optimal case, is associated with a loss of information (unless we remove only redundancies), and therefore when creating a dataset, we always generate a bias. This bias can range from a small, negligible effect to a widespread condition that mischaracterizes the relations present in the larger population and dramatically affects the performance of a model. For this reason, data scientists must pay close attention to how a model is tested, to be sure that new samples are generated by the same process as the training samples were. If there are strong discrepancies, data scientists should warn end users about the differences in the samples.
Since we can assume that similar individuals will behave in a similar way, if the numerosity of the sample set is large enough, we are statistically authorized to draw conclusions that we can extend to the larger, unsampled part of the population. Animals are extremely capable at identifying critical features from a family of samples, and generalizing them to interpret new experiences (for example, a baby learns to distinguish a teddy-bear from a person after only seeing their parents and a few other people). The challenging goal of machine learning is to find the optimal strategies to train models using a limited amount of information, to find all the necessary abstractions that justify their logical processes.
Of course, when we consider our sample populations, we always need to assume that they're drawn from the original data-generating distribution. This isn't a purely theoretical assumption – as we're going to see, if our sample data elements are drawn from a different distribution, the accuracy of our model can dramatically decrease.
For example, if we trained a portrait classifier using 10-megapixel images, and then we used it in an old smartphone with a 1-megapixel camera, we could easily start to find discrepancies in the accuracy of our predictions.
This isn't surprising; many details aren't captured by low-resolution images. You could get a similar outcome by feeding the model with very noisy data sources, whose information content could only be partially recovered.
N values are independent and identically distributed (i.i.d.) if they are sampled from the same distribution, and two different sampling steps yield statistically independent values (that is, p(a, b) = p(a)p(b)). If we sample N i.i.d. values from pdata, we can create a finite dataset X made up of k-dimensional real vectors:

In a supervised scenario, we also need the corresponding labels (with t output values):

When the output has more than two classes, there are different possible strategies to manage the problem. In classical machine learning, one of the most common approaches is One-vs-All, which is based on training N different binary classifiers, where each label is evaluated against all the remaining ones. In this way, N-1 classifications are performed to determine the right class. With shallow and deep neural models, instead, it's preferable to use a softmax function to represent the output probability distribution for all classes:

This kind of output, where zi represents the intermediate values and the sum of the terms is normalized to 1, can be easily managed using the cross-entropy cost function, which we'll discuss in Chapter 2, Loss functions and Regularization. A sharp-eyed reader might notice that calculating the softmax output of a population allows one to obtain an approximation of the data generating process.
This is brilliant, because once the model has been successfully trained and validated with a positive result, it's reasonable to assume that the output corresponding to never-seen samples reflects the real-world joint probability distribution. That means the model has developed an internal representation of the relevant abstractions with a minimum error; which is the final goal of the whole machine learning process.
Before moving on to the discussion of some fundamental preprocessing techniques, it's worth mentioning the problem of domain adaptation, which is one of the most challenging and powerful techniques currently under development.
As discussed, animals can perform abstractions and extend the concepts learned in a particular context to similar, novel contexts. This ability is not only important but also necessary. In many cases, a new learning process could take too long, exposing the animal to all sorts of risks.
Unfortunately, many machine learning models lack this property. They can easily learn to generalize, but always under the condition of coping with samples originating from the same data generating process. Let's suppose that a model M has been optimized to correctly classify the elements drawn from p1(x, y) and the final accuracy is large enough to employ the model in a production environment. After a few tests, a data scientist discovers that p2(x, y) = f(p1(x, y)) is another data generating process that has strong analogies with p1(x, y). Its samples meet the requirements needed to be considered a member of the same global class. For example, p1(x, y) could represent family cars, while p2(x, y) could be a process modeling a set of trucks.
In this case, it's easy to understand that a transformation f(z) is virtually responsible for increasing the size of the vehicles, their relative proportions, the number of wheels, and so on. At this point, can our model M also correctly classify the samples drawn from p2(x, y) by exploiting the analogies? In general, the answer is negative. The observed accuracy decays, reaching the limit of a purely random guess.
The reasons behind this problem are strictly related to the mathematical nature of the models and won't be discussed in this book (the reader who is interested can check the rigorous paper Crammer K., Kearns M., Wortman J., Learning from Multiple Sources, Journal of Machine Learning Research, 9/2008). However, it is helpful to consider such a scenario. The goal of domain adaptation is to find the optimal methods to let a model shift from M to M' and vice versa, in order to maximize its ability to work with a specific data generating process.
It's within the limits of reasonable change, for example, for a component of the model to recognize the similarities between a car and truck (for example, they both have a windshield and a radiator) and force some parameters to shift from their initial configuration, whose targets are cars, to a new configuration based on trucks. This family of methods is clearly more suitable to represent cognitive processes. Moreover, it has the enormous advantage of allowing reuse of the same models for different purposes without the need to re-train them from scratch, which is currently often a necessary condition to achieve acceptable performances.
This topic is still enormously complex; certainly, it's too detailed for a complete discussion in this book. Therefore, unless we explicitly declare otherwise, in this book you can always assume we are working with a single data generating process, from which all the samples will be drawn.
Now, let's introduce some important data preprocessing concepts that will be helpful in many practical contexts.
Scaling datasets
Many algorithms (such as logistic regression, Support Vector Machines (SVMs) and neural networks) show better performances when the dataset has a feature-wise null mean. Therefore, one of the most important preprocessing steps is so-called zero-centering, which consists of subtracting the feature-wise mean Ex[X] from all samples:

This operation, if necessary, is normally reversible, and doesn't alter relationships either among samples or among components of the same sample. In deep learning scenarios, a zero-centered dataset allows us to exploit the symmetry of some activation functions, driving our model to a faster convergence (we're going to discuss these details in the next chapters).
Zero-centering is not always enough to guarantee that all algorithms will behave correctly. Different features can have very different standard deviations, and therefore, an optimization that works considering the norm of the parameter vector (see the section about regularization) will tend to treat all the features in the same way. This equal treatment can produce completely different final effects; features with a smaller variance will be affected more than features with a larger variance.
In a similar way, when single features contribute to finding the optimal parameters, features with a larger variance can take control over the other features, forcing them in the context of the problem to become similar to constant values. In this way, those less-varied features lose the ability to influence the end solution (for example, this problem is a common limiting factor when it comes to regressions and neural networks). For this reason, If 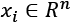 ,
,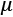 , and
, and 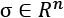 and are computed considering every single feature for the whole dataset, it's often helpful to divide the zero-centered samples by the feature-wise standard deviation, obtaining the so-called z-score:
and are computed considering every single feature for the whole dataset, it's often helpful to divide the zero-centered samples by the feature-wise standard deviation, obtaining the so-called z-score:

The result is a transformed dataset where most of the internal relationships are kept, but all the features have a null mean and unit variance. The whole transformation is completely reversible when it's necessary to remap the vectors onto the original space.
We can now analyze other approaches to scaling that we might choose for specific tasks (for example, datasets with outliers).
Range scaling
Another approach to scaling is to set the range where all features should lie. For example, if 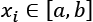 so that
so that 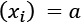 and
and 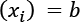 , the transformation will force all the values to lie in a new range
, the transformation will force all the values to lie in a new range 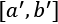 , as shown in the following figure:
, as shown in the following figure:

Schematic representation of a range scaling
Range scaling behaves in a similar way to standard scaling, but in this case, both the new mean and the new standard deviation are determined by the chosen interval. In particular, if the original features have symmetrical distributions, the new standard deviations will be very similar, even if not exactly equal. For this reason, this method can often be chosen as an alternative to a standard scaling (for example, when it's helpful to bound all the features in the range [0, 1]).
Robust scaling
The previous two methods have a common drawback: they are very sensitive to outliers. In fact, when the dataset contains outliers, their presence will affect the computation of both mean and standard deviation, shifting the values towards the outliers. An alternative, robust approach is based on the usage of quantiles. Given a distribution p over a range [a, b], the most common quantile, called median, 50th percentile or second quartile (Q2), is the value the splits the range [a, b] into two subsets so that 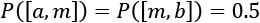 . That is to say, in a finite population, the median is the value in the central position.
. That is to say, in a finite population, the median is the value in the central position.
For example, considering the set A = {1, 2, 3, 5, 7, 9}, we have:

If we add the value 10, the set A, we get 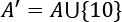 :
:

In a similar way, we can define other percentiles or quantiles. A common choice for scaling the data is the Interquartile Range (IQR), sometimes called H-spread, defined as:

In the previous formula, Q1 is the cut-point the divides the range [a, b] so that 25% of the values are in the subset [a, Q1], while Q2 divides the range so that 75% of the values are in the subset [a, Q2]. Considering the previous set A', we get:

Given these definitions, it's easy to understand that IQR has a low sensitivity to outliers. In fact, let's suppose that a feature lies in the range [-1, 1] without outliers. In a larger dataset, we observe the interval [-2, 3]. If the effect is due to the presence of outliers (for example, the new value 10 added to A), their numerosity is much smaller than the one of normal points, otherwise they are part of the actual distribution. Therefore, we can cut them out from the computation by setting an appropriate quantile. For example, we might want to exclude from our calculations all those features whose probability is lower than 10%. In that case, we would need to consider the 5th and the 95th percentiles in a double-tailed distribution and use their difference QR = 95th – 5th.
Considering the set A', we get IQR = 5.5, while the standard deviation is 3.24. This implies that a standard scaling will compact the values less than a robust scaling. This effect becomes larger and larger as we increase the quantile range (for example, using the 95th and 5th percentiles, 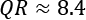 ). However, it's important to remember that this technique is not an outlier filtering method. All the existing values, including the outliers, will be scaled. The only difference is that the outliers are excluded from the calculation of the parameters, and so their influence is reduced, or completely removed.
). However, it's important to remember that this technique is not an outlier filtering method. All the existing values, including the outliers, will be scaled. The only difference is that the outliers are excluded from the calculation of the parameters, and so their influence is reduced, or completely removed.
The robust scaling procedure is very similar to the standard one, and the transformed values are obtained using the feature-wise formula:

Where m is the median and QR is the quantile range (for example, IQR).
Before we discuss other techniques, let's compare these methods using a dataset containing 200 points sampled from a multivariate Gaussian distribution with 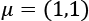 and
and 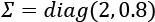 :
:
import numpy as np
nb_samples = 200
mu = [1.0, 1.0]
covm = [[2.0, 0.0], [0.0, 0.8]]
X = np.random.multivariate_normal(mean=mu, cov=covm, size=nb_samples)
At this point, we employ the following scikit-learn classes:
StandardScaler, whose main parameters arewith_meanandwith_std, both Booleans, indicating whether the algorithm should zero-center and whether it should divide by the standard deviations. The default values are bothTrue.MinMaxScaler, whose main parameter isfeature_range, which requires a tuple or list of two elements (a, b) so that a < b. The default value is (0, 1).RobustScaler, which is mainly based on the parameterquantile_range. The default is (25, 75) corresponding to the IQR. In a similar way toStandardScaler, the class accepts the parameterswith_centeringandwith_scaling, that selectively activate/deactivate each of the two functions.
In our case, we're using the default configuration for StandardScaler, feature_range=(-1, 1) for MinMaxScaler, and quantile_range=(10, 90) for RobustScaler:
from sklearn.preprocessing import StandardScaler, RobustScaler, MinMaxScaler
ss = StandardScaler()
X_ss = ss.fit_transform(X)
rs = RobustScaler(quantile_range=(10, 90))
X_rs = rs.fit_transform(X)
mms = MinMaxScaler(feature_range=(-1, 1))
X_mms = mms.fit_transform(X)
The results are shown in the following figure:

Original dataset (top left), range scaling (top right), standard scaling (bottom left), and robust scaling (bottom right)
In order to analyze the differences, I've kept the same scale for all the diagrams. As it's possible to see, the standard scaling performs a shift of the mean and adjusts the points so that it's possible to consider them as drawn from N(0, I). Range scaling behaves in almost the same way and in both cases, it's easy to see how the variances are negatively affected by the presence of a few outliers.
In particular, looking at the result of range scaling, the shape is similar to an ellipse and the roundness—implied by a symmetrical distribution—is obtained by including also the outliers. Conversely, robust scaling is able to produce an almost perfect normal distribution N(0, I) because the outliers are kept out of the calculations and only the central points contribute to the scaling factor.
We can conclude this section with a general rule of thumb: standard scaling is normally the first choice. Range scaling can be chosen as a valid alternative when it's necessary to project the values onto a specific range, or when it's helpful to create sparsity. If the analysis of the dataset has highlighted the presence of outliers and the task is very sensitive to the effect of different variances, robust scaling is the best choice.
Normalization
One particular preprocessing method is called normalization (not to be confused with statistical normalization, which is a more complex and generic approach) and consists of transforming each vector into a corresponding one with a unit norm given a predefined norm (for example, L2):

Given a zero-centered dataset X, containing points 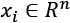 , the normalization using the L2 (or Euclidean) norm transforms each value into a point lying on the surface of a hypersphere with unit radius, and centered in
, the normalization using the L2 (or Euclidean) norm transforms each value into a point lying on the surface of a hypersphere with unit radius, and centered in 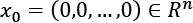 (by definition all the points on the surface have
(by definition all the points on the surface have 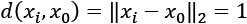 ).
).
Contrary to the other methods, normalizing a dataset leads to a projection where the existing relationships are kept only in terms of angular distance. To understand this concept, let's perform a normalization of the dataset defined in the previous example, using the scikit-learn class Normalizer with the parameter norm='l2':
from sklearn.preprocessing import Normalizer
nz = Normalizer(norm='l2')
X_nz = nz.fit_transform(X)
The result is shown in the following figure:

Normalized bidimensional dataset. All points lie on a unit circle
As we expected, all the points now lie on a unit circle. At this point, the reader might ask how such a preprocessing step could be helpful. In some contexts, such as Natural Language Processing (NLP), two feature vectors are different in proportion to the angle they form, while they are almost insensitive to Euclidean distance.
For example, let's imagine that the previous diagram defines four semantically different concepts, which are located in the four quadrants. In particular, imagine that opposite concepts (for example, cold and warm) are located in opposite quadrants so that the maximum distance is determined by an angle of 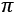 radians (180°). Conversely, two points whose angle is very small can always be considered similar.
radians (180°). Conversely, two points whose angle is very small can always be considered similar.
In this common case, we assume that the transition between concepts is semantically smooth, so two points belonging to different sets can always be compared according to their common features (for example, the boundary between warm and cold can be a point whose temperature is the average between the two groups). The only important thing to know is that if we move along the circle far from a point, increasing the angle, the dissimilarity increases. For our purposes, let's consider the points (-4, 0) and (-1, 3), which are almost orthogonal in the original distribution:
X_test = [
[-4., 0.],
[-1., 3.]
]
Y_test = nz.transform(X_test)
print(np.arccos(np.dot(Y_test[0], Y_test[1])))
The output of the previous snippet is:
1.2490457723982544
The dot product between two vectors x1 and x2 is equal to:

The last step derives from the fact that both vectors have unit norms. Therefore, the angle they form after the projection is almost 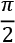 , indicating that they are indeed orthogonal. If we multiply the vectors by a constant, their Euclidean distance will obviously change, but the angular distance after normalization remains the same. I invite you to check it!
, indicating that they are indeed orthogonal. If we multiply the vectors by a constant, their Euclidean distance will obviously change, but the angular distance after normalization remains the same. I invite you to check it!
Therefore, we can completely get rid of the relative Euclidean distances and work only with the angles, which, of course, must be correlated to an appropriate similarity measure.
Whitening
Another very important preprocessing step is called whitening, which is the operation of imposing an identity covariance matrix to a zero-centered dataset:

As the covariance matrix 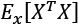 is real and symmetrical, it's possible to eigendecompose it without the need to invert the eigenvector matrix:
is real and symmetrical, it's possible to eigendecompose it without the need to invert the eigenvector matrix:

The matrix V contains the eigenvectors as columns, and the diagonal matrix 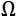 contains the eigenvalues. To solve the problem, we need to find a matrix A, such that:
contains the eigenvalues. To solve the problem, we need to find a matrix A, such that:

Using the eigendecomposition previously computed, we get:

Hence, the matrix A is:

One of the main advantages of whitening is the decorrelation of the dataset, which allows for an easier separation of the components. Furthermore, if X is whitened, any orthogonal transformation induced by the matrix P is also whitened:

Moreover, many algorithms that need to estimate parameters that are strictly related to the input covariance matrix can benefit from whitening, because it reduces the actual number of independent variables. In general, these algorithms work with matrices that become symmetrical after applying the whitening.
Another important advantage in the field of deep learning is that the gradients are often higher around the origin and decrease in those areas where the activation functions (for example, the hyperbolic tangent or the sigmoid) saturate (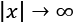 ). That's why the convergence is generally faster for whitened—and zero-centered—datasets.
). That's why the convergence is generally faster for whitened—and zero-centered—datasets.
In the following graph, it's possible to compare an original dataset and the result of whitening, which in this case is both zero-centered and with an identity covariance matrix:

Original dataset (left) and whitened version (right)
When a whitening process is needed, it's important to consider some important details. The first one is that there's a scale difference between the real sample covariance and the estimation 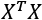 , often adopted with the Singular Value Decomposition (SVD). The second one concerns some common classes implemented by many frameworks, such as scikit-learn's
, often adopted with the Singular Value Decomposition (SVD). The second one concerns some common classes implemented by many frameworks, such as scikit-learn's StandardScaler. In fact, while zero-centering is a feature-wise operation, a whitening filter needs to be computed considering the whole covariance matrix; StandardScaler implements only unit variance and feature-wise scaling.
Luckily, all scikit-learn algorithms that can benefit from a whitening preprocessing step provide a built-in feature, so no further actions are normally required. However, for all readers who want to implement some algorithms directly, I've written two Python functions that can be used for both zero-centering and whitening. They assume a matrix X with a shape (NSamples × n). In addition, the whiten() function accepts the parameter correct, which allows us to apply the scaling correction. The default value for correct is True:
import numpy as np
def zero_center(X):
return X - np.mean(X, axis=0)
def whiten(X, correct=True):
Xc = zero_center(X)
_, L, V = np.linalg.svd(Xc)
W = np.dot(V.T, np.diag(1.0 / L))
return np.dot(Xc, W) * np.sqrt(X.shape[0]) if correct else 1.0
Training, validation, and test sets
As we have previously discussed, the numerosity of the sample available for a project is always limited. Therefore, it's usually necessary to split the initial set X, together with Y, each of them containing N i.i.d. elements sampled from pdata, into two or three subsets as follows:
- Training set used to train the model
- Validation set used to assess the score of the model without any bias, with samples never seen before
- Test set used to perform the final validation before moving to production
The hierarchical structure of the splitting process is shown in the following figure:

Hierarchical structure of the process employed to create training, validation, and test sets
Considering the previous diagram, generally, we have:

The sample is a subset of the potential complete population, which is partially inaccessible. Because of that, we need to limit our analysis to a sample containing N elements. The training set and the validation/test set are disjoint (that is, the evaluation is carried out using samples never seen during the training phase).
The test set is normally obtained by removing Ntest samples from the initial validation set and keeping them apart until the final evaluation. This process is quite straightforward:
- The model M is trained using the training set
- M is evaluated using the validation set and a designated Score(•) function
- If Score(M) > Desired accuracy:
perform the final test to confirm the results
- Otherwise, the hyperparameters are modified and the process restarts
Since the model is always evaluated on samples that were not employed in the training process, the Score(•) function can determine the quality of the generalization ability developed by the model. Conversely, an evaluation performed using the training sample can help us understand whether the model is basically able to learn the structure of the dataset. We'll discuss these concepts further over the next few sections.
The choice of using two (training and validation) or three (training, validation, and test) sets is normally related to the specific context. In many cases, a single validation set, which is often called the test set, is used throughout the whole process. That's usually because the final goal is to have a reliable set of i.i.d. elements that will never be employed for training and, consequently, whose prediction results reflect the unbiased accuracy of the model. In this book, we'll always adopt this strategy, using the expression test set instead of validation set.
Depending on the nature of the problem, it's possible to choose a split percentage ratio of 70% – 30%, which is a good practice in machine learning, where the datasets are relatively small, or a higher training percentage of 80%, 90%, or up to 99% for deep learning tasks where the numerosity of the samples is very high. In both cases, we're assuming that the training set contains all the information we'll require for a consistent generalization.
In many simple cases, this is true and can be easily verified; but with more complex datasets, the problem becomes harder. Even if we draw all the samples from the same distribution, it can happen that a randomly selected test set contains features that are not present in other training samples. When this happens, it can have a very negative impact on global accuracy and, without other methods, it can also be very difficult to identify.
This is one of the reasons why, in deep learning, training sets are huge: considering the complexity of the features and structure of the data generating the distributions, choosing large test sets can limit the possibility of learning particular associations. This is a consequence of an effect called overfitting, which we'll discuss later in this chapter.
In scikit-learn, it's possible to split the original dataset using the train_test_split() function, which allows specifying the train/test size, and if we expect to have randomly shuffled sets (which is the default). For example, if we want to split X and Y, with 70% training and 30% test, we can use:
from sklearn.model_selection import train_test_split
X_train, X_test, Y_train, Y_test = train_test_split(X, Y, train_size=0.7, random_state=1000)
Shuffling the sets is always good practice, in order to reduce the correlation between samples (the method train_test_split has a parameter called shuffle that allows this to be done automatically). In fact, we have assumed that X is made up of i.i.d samples, but often two subsequent samples have a strong correlation, which reduces the training performance. In some cases, it's also useful to re-shuffle the training set after each training epoch; however, in the majority of our examples, we'll work with the same shuffled dataset throughout the whole process.
Shuffling has to be avoided when working with sequences and models with memory. In all those cases, we need to exploit the existing correlation to determine how the future samples are distributed. Whenever an additional test set is needed, it's always possible to reuse the same function: splitting the original test set into a larger component, which becomes the actual validation set, and a smaller one, the new test set that will be employed for the final performance check.
When working with NumPy and scikit-learn, it's always a good practice to set the random seed to a constant value, so as to allow other people to reproduce the experiment with the same initial conditions. This can be achieved by calling np.random.seed(...) and using the random-state parameter present in many scikit-learn methods.
Cross-validation
A valid method to detect the problem of wrongly selected test sets is provided by the cross-validation (CV) technique. In particular, we're going to use the K-Fold cross-validation approach. The idea is to split the whole dataset X into a moving test set and a training set made up of the remaining part. The size of the test set is determined by the number of folds, so that during k iterations, the test set covers the whole original dataset.
In the following diagram, we see a schematic representation of the process:

K-Fold cross-validation schema
In this way, we can assess the accuracy of the model using different sampling splits, and the training process can be performed on larger datasets; in particular, on (k-1)N samples. In an ideal scenario, the accuracy should be very similar in all iterations; but in most real cases, the accuracy is quite below average.
This means that the training set has been built excluding samples that contain all the necessary examples to let the model fit the separating hypersurface considering the real pdata. We're going to discuss these problems later in this chapter. However, if the standard deviation of the accuracies is too large—a threshold must be set according to the nature of the problem/model—that probably means that X hasn't been drawn uniformly from pdata, and it's useful to evaluate the impact of the outliers in a preprocessing stage. In the following graph, we see the plot of 15-fold CV performed on a Logistic Regression:

Cross-validation accuracies
The values oscillate from 0.84 to 0.95, with an average of 0.91, marked on the graph as a solid horizontal line. In this particular case, considering the initial purpose was to use a linear classifier, we can say that all folds yield high accuracies, confirming that the dataset is linearly separable; however, there are some samples, which were excluded in the ninth fold, that are necessary to achieve a minimum accuracy of about 0.88.
K-Fold cross-validation has different variants that can be employed to solve specific problems:
- Stratified K-Fold: A standard K-Fold approach splits the dataset without considering the probability distribution
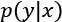 , therefore some folds may theoretically contain only a limited number of labels. Stratified K-Fold, instead, tries to split X so that all the labels are equally represented.
, therefore some folds may theoretically contain only a limited number of labels. Stratified K-Fold, instead, tries to split X so that all the labels are equally represented. - Leave-one-out (LOO): This approach is the most drastic because it creates N folds, each of them containing N-1 training samples and only one test sample. In this way, the maximum possible number of samples is used for training, and it's quite easy to detect whether the algorithm is able to learn with sufficient accuracy, or if it's better to adopt another strategy.
- The main drawback of this method is that N models must be trained, and when N is very large this can cause a performance issue. It's also an issue that with a large number of samples, the probability that two random values are similar increases, and therefore many of the folds will yield almost identical results. At the same time, LOO limits the possibilities for assessing the generalization ability of a model, because a single test sample is not enough for a reasonable estimation.
- Leave-P-out (LPO): In this case, the number of test samples is set to p non-disjoint sets, so the number of folds is equal to the binomial coefficient of n over p. This approach mitigates LOO's drawbacks, and it's a trade-off between K-Fold and LOO. The number of folds can be very high, but it's possible to control it by adjusting the number p of test samples; however, if p isn't small or big enough, the binomial coefficient can exponentially explode, as shown in the following figure in case of n=20 and
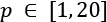 :
:

Exploding effect of the binomial coefficient when p is about half of n
Scikit-learn implements all those methods, with some other variations, but I suggest always using the cross_val_score() function, which is a helper that allows applying the different methods to a specific problem. It uses Stratified K-Fold for categorical classifications and Standard K-Fold for all other cases. Let's now try to determine the optimal number of folds, given a dataset containing 500 points 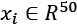 with redundancies, internal non-linearities, and belonging to 5 classes:
with redundancies, internal non-linearities, and belonging to 5 classes:
from sklearn.datasets import make_classification
from sklearn.preprocessing import StandardScaler
X, Y = make_classification(n_samples=500, n_classes=5,
n_features=50, n_informative=10,
n_redundant=5, n_clusters_per_class=3,
random_state=1000)
ss = StandardScaler()
X = ss.fit_transform(X)
As the first exploratory step, let's plot the learning curve using a Stratified K-Fold with 10 splits; this assures us that we'll have a uniform class distribution in every fold:
import numpy as np
from sklearn.linear_model import LogisticRegression
from sklearn.model_selection import learning_curve, StratifiedKFold
lr = LogisticRegression(solver='lbfgs', random_state=1000)
splits = StratifiedKFold(n_splits=10, shuffle=True, random_state=1000)
train_sizes = np.linspace(0.1, 1.0, 20)
lr_train_sizes, lr_train_scores, lr_test_scores = \
learning_curve(lr, X, Y, cv=splits, train_sizes=train_sizes,
n_jobs=-1, scoring='accuracy',
shuffle=True, random_state=1000)
The result is shown in the following diagram:

Learning curves for a Logistic Regression classification
The training curve decays when the training set size reaches its maximum, and converges to a value slightly larger than 0.6. This behavior indicates that the model is unable to fully capture the dynamics of X, and it has good performances only when the training set size is very small (that is, the actual data generating process is not fully covered). Conversely, the test performances improve when the training set is larger. This is an obvious consequence of the wider experience that the classifier gains when more and more points are employed.
Considering both the training and test accuracy trends, we can conclude that in this case a training set larger than about 270 points doesn't yield any strong benefit. On the other hand, since the test accuracy is extremely important, it's preferable to use the maximum number of points. As we're going to discuss later in this chapter, it indicates how well the model generalizes. In this case, the average training accuracy is worse, but there's a small benefit in the test accuracy. I've chosen this example because it's a particular case that requires a trade-off. In many cases, the curves grow proportionally, and determining the optimal number of folds is straightforward.
However, when the problem is harder, as it is in this case—considering the nature of the classifier—the choice is not obvious, and analyzing the learning curve becomes an indispensable step. Before we move on, we can try to summarize the rule. We need to find the optimal number of folds so that cross-validation guarantees an unbiased measure of the performances.
As a dataset X is drawn from an underlying data generating process, the amount of information that X carries is bounded by pdata. This means that an increase of the dataset's size over a certain threshold can only introduce redundancies, which cannot improve the performance of the model. The optimal number of folds, or the size of the folds, can be determined by considering the point at which both training and test average accuracies stabilize. The corresponding training set size allows us to use the largest possible test sample size for performance evaluations. Let's now compute the average CV accuracies for a different number of folds:
import numpy as np
from sklearn.linear_model import LogisticRegression
from sklearn.model_selection import cross_val_score
mean_scores = []
cvs = [x for x in range(5, 100, 10)]
for cv in cvs:
score = cross_val_score(LogisticRegression(solver='lbfgs',
random_state=1000),
X, Y, scoring='accuracy', n_jobs=-1,
cv=cv)
mean_scores.append(np.mean(score))
The result is shown in the following figure:

Average cross-validation accuracy for a different number of folds
The curve has a peak corresponding to 15-fold CV, which corresponds to a training set size of 466 points. In our previous analysis, we have discovered that such a value is close to the optimal one. On the other side, a larger number of folds implies smaller test sets.
We have seen that the average CV accuracy depends on a trade-off between training and test set sizes. Therefore, when the number of folds increases, we should expect an improvement in the performances. This result becomes clear with 85 folds. In this case, only 6 samples are used for testing purposes (1.2%), which means the validation is not particularly reliable, and the average value is associated with a very large variance (that is, in some lucky cases, the CV accuracy can be large, while in the remaining ones, it can be close to 0).
Considering all the factors, the best choice remains k=15, which implies the usage of 34 test samples (6.8%). I hope it's clear the right choice of k is a problem itself; however, in practice, a value in the range [5, 15] is often the most reasonable default choice. The goal of a good choice is also to maximize the stochasticity of CV and, consequently, to reduce the cross-correlations between estimations. Very small folds imply that many models are highly correlated, while over-large folds reduce the learning ability of the model. Therefore, a good trade-off should never prefer either very small values (acceptable only if the dataset is extremely small) nor over-large ones.
Of course, this value is strictly correlated to the nature of the task and to the structure of the dataset. In some cases, just 3 to 5% of test points can be enough to perform a correct assessment; in many other ones, a larger set is needed in order to capture the dynamics of all regions.
As a general rule, I always encourage the employment of CV for performance measurements. The main drawback of this method is its computational complexity. In the context of deep learning, for example, a training process can require hours or days, and repeating it without any modification of the hyperparameters can be unacceptable. In all these cases, a standard training-test set decomposition will be used, assuming that for both sets the numerosity is large enough to guarantee full coverage of the underlying data generating process.






